K-merとは? DNA/アミノ酸配列の部分配列のこと
k-merとは?
一言でいうと、「 長さkのDNAの部分配列のこと 」である。
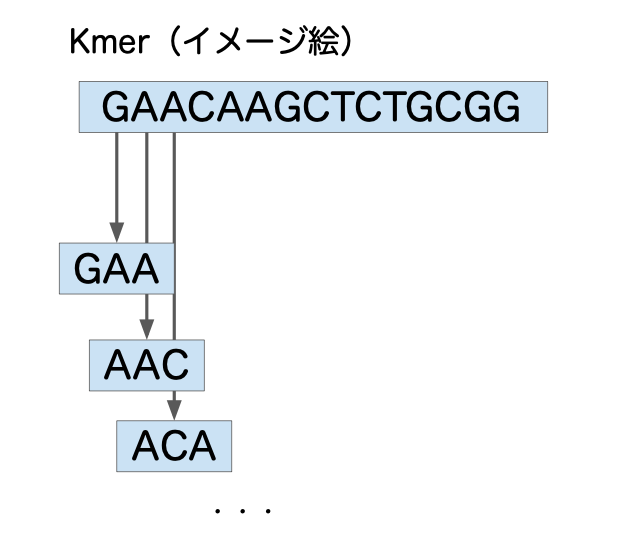
例えば「GAACAAGCTCTGCGG」という配列があった場合、この配列の3-merは、
GAA, AAC, ACA, CAA, …. CGG となる。
図示すると以下となる。
k-merの活用方法
現状では大きくわけて以下の3つの活用方法がある。
-
配列検索のためのフィルタリング
-
de Brujinグラフによるゲノムアセンブリ
-
種の判別
# 配列検索のためのフィルタリング
BLASTなどの配列検索プログラムでは、配列の一部分が完全一致する部分を探し、そこからアライメントを開始する。
クエリ配列 => GAACAAGCTCTGCGG
DB配列 => CAATAACTCTACGAA
という配列をアライメントする際、
クエリ配列を k-merに分割する。(ここでは4-merに分割する。)
GAAC, AACA, ACAA, CAAG, …. GCGG
そして、これら4-merと一致する部分からアライメントを始めることとなる。
今回の場合、CTCT(クエリの8塩基目)からアライメントを始めることとなる。

こうすることで、アライメント領域の大幅な節約ができる。
なお、BLASTのデフォルトでは、DNAでは11-merを、アミノ酸では3-merを使用する。
# de Brujinグラフによるゲノムアセンブリ
以下のリンクなどに詳しい。そちらを参照されたし。
https://hoxo-m.hatenablog.com/entry/20100930/p1
# 種の判別
以下の2つのゲノム配列の類似度を求めたいとします。
ゲノムA:GAACAAGCTCTGCGG
ゲノムB:CAATAACTCTACGAA
通常ならばアライメントを取るだけですが、このゲノムの3-merをプロファイルを求めてみます。
| A | B | |
|---|---|---|
| AAC | 1.0 | 1.0 |
| AAG | 1.0 | 0.0 |
| AAT | 0.0 | 1.0 |
| ACA | 1.0 | 0.0 |
| ACG | 0.0 | 1.0 |
| ACT | 0.0 | 1.0 |
| AGC | 1.0 | 0.0 |
| ATA | 0.0 | 1.0 |
| CAA | 1.0 | 1.0 |
| CGA | 0.0 | 1.0 |
| CGG | 1.0 | 0.0 |
| CTA | 0.0 | 1.0 |
| CTC | 1.0 | 1.0 |
| CTG | 1.0 | 0.0 |
| GAA | 1.0 | 1.0 |
| GCG | 1.0 | 0.0 |
| GCT | 1.0 | 0.0 |
| TAA | 0.0 | 1.0 |
| TAC | 0.0 | 1.0 |
| TCT | 1.0 | 1.0 |
| TGC | 1.0 | 0.0 |
そしてこのk-merのプロファイルを比較することで擬似的に配列同士の類似度を算出することができます。
BLASTなどの配列アライメントツールは基本的に遅いため、大量にアライメントを取る必要があるときは、Kmerによる簡易な比較を用いられることもあります。