Qiime2 チュートリアル
Qiime2 チュートリアル
データを得てからQiime2にてtsvファイルにて出力するまでの流れを説明していきます。
前提知識
本チュートリアルにおける前提知識は以下とします。
- 基礎的なシェルコマンドを理解していること
- vim または emacsでlinuxにてファイル編集ができること
基礎的なシェルコマンドについて学びたい場合、以下のサイト等で学ぶことができます。
(基本的なシェルコマンド)

シェルコマンド入門編1:シェルコマンドの基本を身に付けようのチャプター一覧
(Vim)
vim入門 (全18回) - プログラミングならドットインストール
準備(サーバーの準備〜データダウンロードまで)
色々と準備します。具体的には、以下の3つを準備します。
- 解析環境(サーバー)
- Qiime2のインストール
- 解析データのダウンロード
①サーバー環境構築
16S rRNA解析ですが、ある程度の計算力のあるPCが必要になってきます。一応ローカルPCでの実行も可能なくらいの計算量ではあります。実際問題、アカデミア所属の方ならば遺伝研のサーバーが無料なので、そちらを借りるのがよいと思います。
企業の方はVPSを借りるか、ローカルのPCで解析がよいかと思います。今回私はローカル環境で解析を行っています。メモリがボトルネックとなることが多いです。メモリ4GBくらい欲しい。
【遺伝研のHP】
遺伝研スーパーコンピュータシステム | NIG supercomputer
【ConohaのHP】

レンタルサーバーならConoHa|登録者数50万アカウント突破
Conohaでの環境構築は以下に記載しました。
404 Error - Not Found
②Qiime2の環境構築
Qiime2のインストールを行います。基本的には公式のページの通りにインストールしていきます。
Qiime2のバージョンとしては、2022年2月版を用います。最新版でない場合、最新版でないけど問題ないか?という質問が出てきますが、今回のバージョンで動くコマンドが知りたいので、問題ないと選んでください。
【公式ページ】
QIIME 2 user documentation — QIIME 2 2022.11.1 documentation
## Qiime2のインストール方法について
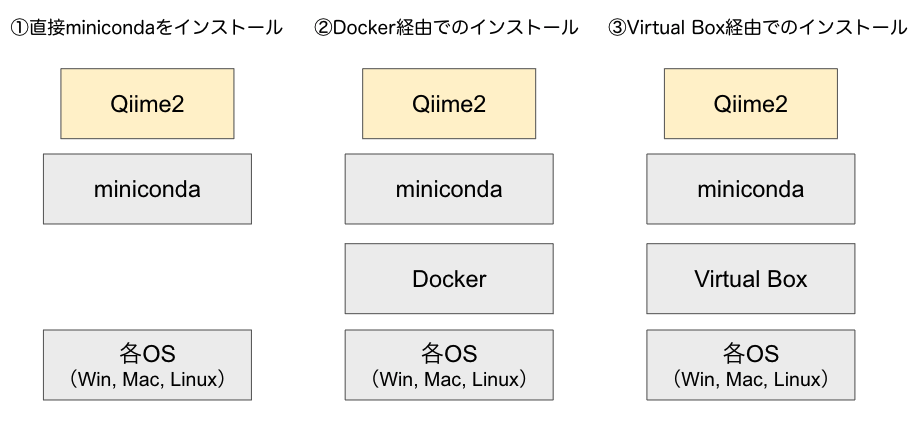
公式ページにも基本的に3通りの方法を準備してくれています。いずれの方法でもよいのですが、公式の推奨に従い、①直接minicondaをインストールをしていきます。
## minicondaのインストール
【minicondaとは?】
本来、Python向けの環境構築ソフトです。
- Conda環境は独立したPythonの実行環境で、他の環境に影響を与えずにPythonのバージョンを用途によって切り替えたり、パッケージをインストールしたりできます。
Conda コマンド - python.jp
【minicondaの公式ページ】
Miniconda — conda documentation
【minicondaのインストールコマンド】
シェルをダウンロードしてきて、実行すれば終わります。
# ダウンロードをコマンドで行っています。
# OSによって微妙にURLが違うので注意してください。
curl -o miniconda.sh https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash miniconda.sh
# 問答は以下でOK
# 最初:qを押す(qを押さないと規約が読み終わらない)
# 1コ目:インストールしてよいか? => yes
# 2コ目:どこにインストールする? => (デフォルト;何も入力しない)
# 3コ目:yes => (デフォルト) => yes
source ~/.bashrc
rm miniconda.sh
③Qiime2のインストール
公式ページに従ってインストールしていきます。
【公式ページ】
Natively installing QIIME 2 — QIIME 2 2022.2.0 documentation
【コマンド】
# 環境構築用のテンプレファイルのダウンロード。
wget https://data.qiime2.org/distro/core/qiime2-2022.2-py38-linux-conda.yml
# 環境構築を行う。長い。
conda env create -n qiime2-2022.2 --file qiime2-2022.2-py38-linux-conda.yml
rm qiime2-2022.2-py38-linux-conda.yml
conda activate qiime2-2022.2
qiime —help とコマンドを打って、色々な情報が帰ってきたらOKです。
④データのダウンロード
実際は参加されているプロジェクトごとにデータがあると思います。本プロジェクトではDRAにて公開されているデータをダウンロードしたいと思います。
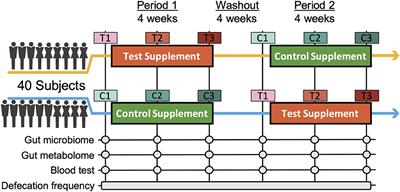
The Nutritional Efficacy of Chlorella Supplementation Depends on the Individual Gut Environment: A Randomised Control Study
【コマンド】
wget https://ddbj.nig.ac.jp/public/ddbj_database/dra/fastq/DRA010/DRA010607/DRX229627/DRR239752_1.fastq.bz2
wget https://ddbj.nig.ac.jp/public/ddbj_database/dra/fastq/DRA010/DRA010607/DRX229627/DRR239752_2.fastq.bz2
wget https://ddbj.nig.ac.jp/public/ddbj_database/dra/fastq/DRA010/DRA010607/DRX229628/DRR239753_1.fastq.bz2
wget https://ddbj.nig.ac.jp/public/ddbj_database/dra/fastq/DRA010/DRA010607/DRX229628/DRR239753_2.fastq.bz2
以下からダウンロードすることもできます。
Qiime2によるシーケンスデータの解析
シーケンスデータの解析を行っていきます。
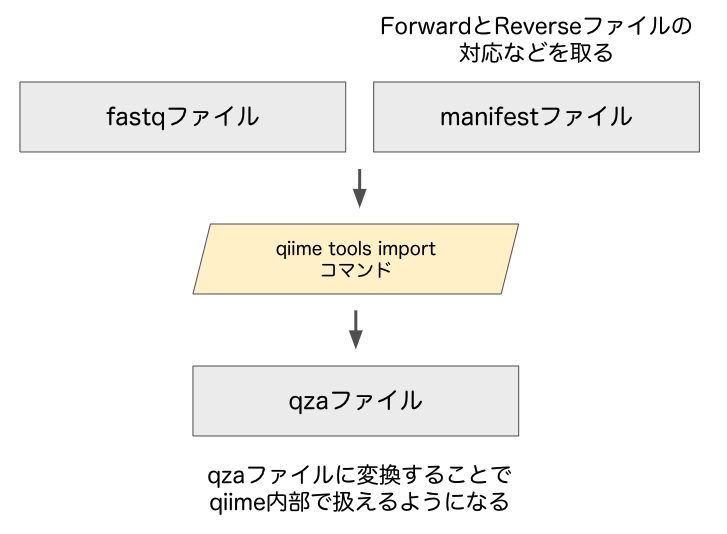
# はじめに(全体概要フローチャート)
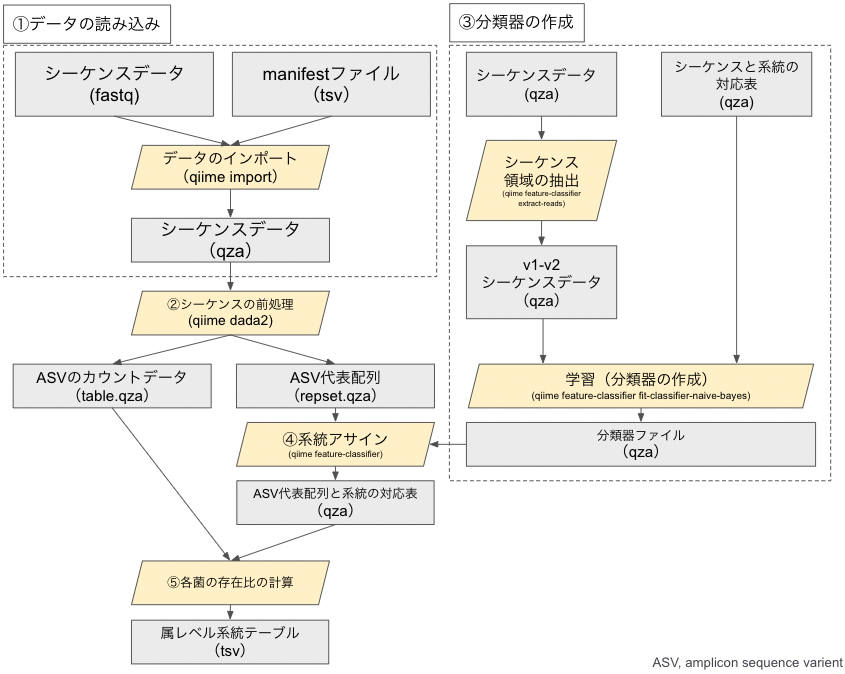
最初に、解析におけるフローチャートを示します。四角部分はファイル、平行四辺形の部分は行うことを表します。
大きく分けて、以下の5ステップがあります。
- データの読み込み
- シーケンスの前処理
- 分類器の作成
- 系統アサイン
- 各菌の存在比の計算
①:データの読み込み
qiime内部で扱うために、 qiime tools import コマンドにてimportをします。importのためには、「fastqファイル」と「manifestファイル」が必要です。まず、fastqファイルを準備します。
## Fastqファイルの準備
今回、home下に以下のようにfastqファイルを配置していきます。(普段使用する際はgzファイル形式の方が多いと思うので、gzに再圧縮しています。コマンドはこのあと示します)
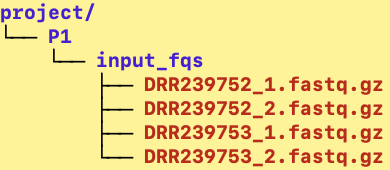
home下にログインした後、以下のようにコマンドを打ってください。
# bash
mkdir -p project/P1/input_fqs
cd project/P1/input_fqs
# データをダウンロード
wget https://ddbj.nig.ac.jp/public/ddbj_database/dra/fastq/DRA010/DRA010607/DRX229627/DRR239752_1.fastq.bz2
wget https://ddbj.nig.ac.jp/public/ddbj_database/dra/fastq/DRA010/DRA010607/DRX229627/DRR239752_2.fastq.bz2
wget https://ddbj.nig.ac.jp/public/ddbj_database/dra/fastq/DRA010/DRA010607/DRX229628/DRR239753_1.fastq.bz2
wget https://ddbj.nig.ac.jp/public/ddbj_database/dra/fastq/DRA010/DRA010607/DRX229628/DRR239753_2.fastq.bz2
# データを解凍
bunzip2 *.bz2
# データを圧縮(fastq => fastq.gz)
gzip *.fastq
## Manifestファイルの構造
Manifestファイルですが、forwardとreverse両方から読み込んでいる場合、以下のような対応表をcsv形式(列の境目をカンマで区切る形式)で作成してください。
【manifest.csvの中身】
sample-id,absolute-filepath,direction
DRR239752,$HOME/project/P1/input_fqs/DRR239752_1.fastq.gz,forward
DRR239752,$HOME/project/P1/input_fqs/DRR239752_2.fastq.gz,reverse
DRR239753,$HOME/project/P1/input_fqs/DRR239753_1.fastq.gz,forward
DRR239753,$HOME/project/P1/input_fqs/DRR239753_2.fastq.gz,reverse
中身はそれぞれ以下のようになっています。
【Manifestファイルの構造に関する説明】

【manifestファイル作成に関する公式ページ】
Importing data — QIIME 2 2022.2.0 documentation
そしたらあとは qiime tools import コマンドにて読み込むことができます。以下のコマンドをうち、sequence.qzaができていれば完了です。
【コマンド】
qiime tools import \
--type SampleData[PairedEndSequencesWithQuality] \
--input-path manifest.csv \
--input-format PairedEndFastqManifestPhred33 \
--output-path sequence.qza
# ls sequence.qza # qzaファイルができていればOK
②:シーケンスの前処理
16S配列の処理を進めます。ここでは、以下の処理を同時に進めます。ただ、3’と5’末端の塩基除去のためにクオリティの確認を先に行います。
- 3’と5’末端の塩基除去
- クオリティフィルタリング
- PhiX由来の配列の除去
- デノイジング
- ペアエンドリードのマージ
- キメラ配列の除去
## クオリティの確認
まず、シーケンスデータのクオリティの確認を行います。クオリティとは、シーケンサーが塩基を読み取ったときにその正確度を算出したスコアです。以下のサイトなどをご参照ください。
クオリティスコア | シークエンサーが出力するクオリティスコアとシーケンシングエラー率
以下のコマンドを叩くことで、 demux.qzvができます。これをダウンロードしてクオリティの確認をします。
qiime demux summarize --i-data sequence.qza --o-visualization demux.qzv
データをダウンロードします。scpコマンドでダウンロードすることができます。
【コマンド@ローカル】
# アカウントやIPアドレスは人によって異なるので変更の必要あり
scp nsmt@0.0.0.0:/home/nsmt/project/P1/demux.qzv ./
【qzvファイルを見るためのページ】
QIIME 2 View
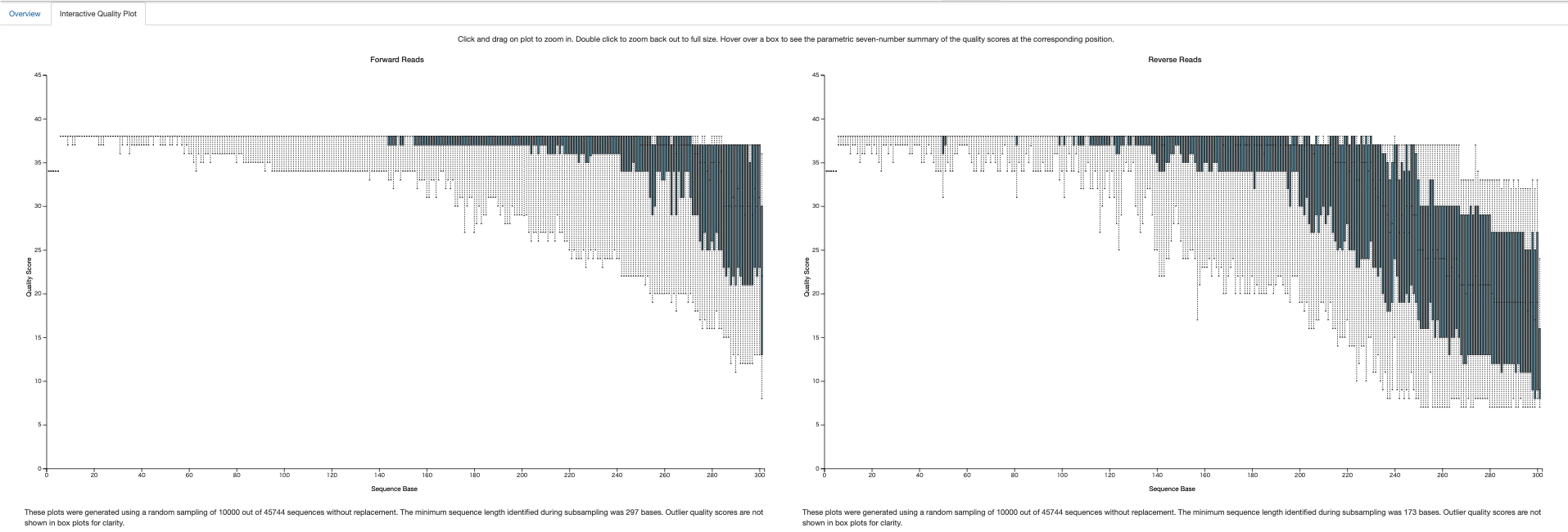
トップページの”Drag and drop or click here”と記載のあるところにアップロードすると、ファイルを見ることができます。今回みたいのはInteractive Quality Plot。以下のような状態が確認できればOKです。大体クオリティ25超えてればOK。
## 16S配列の前処理
クオリティを確認したので、16S配列の前処理を行っていきます。概ねデフォルトで問題ないのですが、3’と5’末端の塩基の長さはデータに応じて指定する必要があります。
| 方向 | 対応 |
|---|---|
| 5'末端 | 0でもよい。プライマー配列などがついている場合はその長さにする。 |
| 3'末端 | クオリティが低いところで切る。ただし、短すぎるとForwardとReverseがつながらない(20塩基以上は塩基が重複していないとツールがエラーを吐く)ため、一定程度の長さにする。長さを変えたことで論文的にツッコミを受けたことは無いが、理論的なベストを探索する場合はFigaroなどを検討する。 |
今回は、5’末端はプライマーの長さに応じて20塩基、19塩基としました。
3’末端はクオリティ値を元に240塩基、または140塩基で切りました。
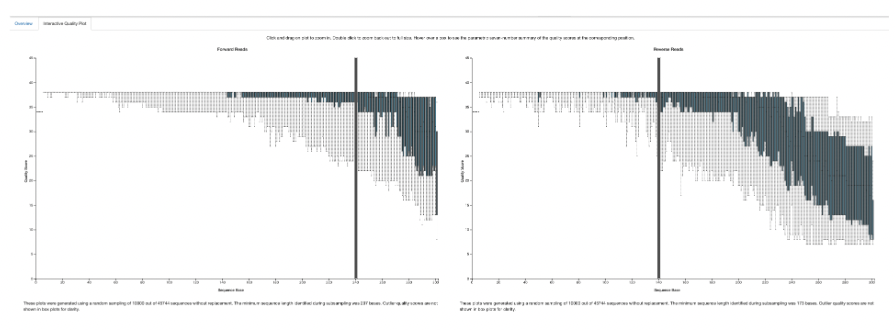
(太い線の所で切っています)
【コマンド】
# ここはデータに応じて変える必要があります!!
# 5'末端はプライマーの長さに応じて20塩基、19塩基
# 3'末端はクオリティ値を元に240塩基、140塩基
qiime dada2 denoise-paired \
--i-demultiplexed-seqs sequence.qza \
--p-trim-left-f 20 \
--p-trim-left-r 19 \
--p-trunc-len-f 240 \
--p-trunc-len-r 140 \
--o-representative-sequences repset.qza \
--o-table table.qza \
--o-denoising-stats denoising_stats.qza
ここで得られる repset.qza の中にASV代表配列が入っています(OTU配列ということもあります)。
詳細な説明は省きますが、このQiime1とQiime2の一番違うところと言われています。以下などに説明あり。
③:分類器の作成
この後、上記で得られたASV配列に対して属や種名をアサインするために、データベースのカスタマイズおよび分類器の作成をします。概要としては、以下のようなことを行います。
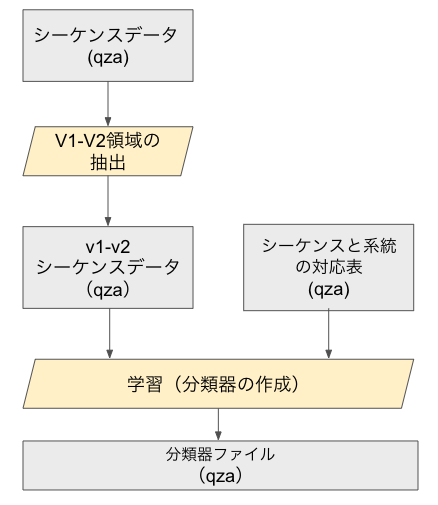
シーケンスデータ、シーケンスと系統の対応表(qzaファイル)は以下から取得することができます。
# データのダウンロード
wget https://data.qiime2.org/2022.2/common/silva-138-99-seqs.qza
wget https://data.qiime2.org/2022.2/common/silva-138-99-tax.qza
# v1-v2領域の抽出
# v1-v2領域は27F-338Rでおよそ300bpのため、極端に長い配列や短い配列は除外する。
qiime feature-classifier extract-reads \
--i-sequences silva-138-99-seqs.qza \
--p-f-primer AGRGTTTGATYMTGGCTCAG \
--p-r-primer TGCTGCCTCCCGTAGGAGT \
--p-min-length 100 \
--p-max-length 600 \
--o-reads silva-138-99-seqs.v1v2.qza
# 学習(分類器の作成)
qiime feature-classifier fit-classifier-naive-bayes \
--i-reference-reads silva-138-99-seqs.v1v2.qza \
--i-reference-taxonomy silva-138-99-tax.qza \
--o-classifier clf.silva-138-99-seqs.v1v2.qza
④:系統アサイン
DADA2によって得られた代表配列に対して属名のアサインを行っていきます。以下のコマンドで行うことができます。
# ASV配列に対して系統アサインを行う
qiime feature-classifier classify-sklearn \
--i-classifier clf.silva-138-99-seqs.v1v2.qza \
--i-reads repset.qza \
--output-dir Taxonomy
⑤:各菌の存在比の計算
代表配列に対して属名のアサインが終わったので、各ASV配列の数を数えることで、どの菌がどのくらいいるか?を出力することができます。以下はQiitaの記事を編集しながら持ってきました。
# ASV代表配列に属するリードの数を元に、
# どの菌がどのくらいいるかを計算
# p-levelを変えるとアサインするレベルを変えられます(例えば、7なら種がでます)
qiime taxa collapse \
--i-table table.qza \
--i-taxonomy Taxonomy/classification.qza \
--p-level 6 \
--o-collapsed-table collapsed_table.genus.qza
# Convert to relative abundance
qiime feature-table relative-frequency \
--i-table collapsed_table.genus.qza \
--o-relative-frequency-table collapsed_table.genus.rel.qza
# Export to biom
qiime tools export \
--input-path collapsed_table.genus.rel.qza \
--output-path exported-feature-table_genus
# Convert biom to tsv
biom convert -i exported-feature-table_genus/feature-table.biom -o genus.tsv --to-tsv --header-key taxonomy
# 出力結果の確認
以下のコマンドで出力結果を確認します。
head genus.tsv
# Constructed from biom file
#OTU ID DRR239752 DRR239753 taxonomy
d__Bacteria;p__Bacteroidota;c__Bacteroidia;o__Bacteroidales;f__Bacteroidaceae;g__Bacteroides 0.3858275826552683 0.23743510142910668
d__Bacteria;p__Actinobacteriota;c__Actinobacteria;o__Bifidobacteriales;f__Bifidobacteriaceae;g__Bifidobacterium 0.05582449273869605 0.23181501900123105
d__Bacteria;p__Bacteroidota;c__Bacteroidia;o__Bacteroidales;f__Rikenellaceae;g__Alistipes 0.05191059841384282 0.030509018894181877
d__Bacteria;p__Firmicutes;c__Clostridia;o__Lachnospirales;f__Lachnospiraceae;g__[Eubacterium]_ventriosum_group 0.021062931300854876 0.02210565754964406
d__Bacteria;p__Proteobacteria;c__Gammaproteobacteria;o__Burkholderiales;f__Sutterellaceae;g__Sutterella 0.01658255227108868 0.02317614944066799
d__Bacteria;p__Actinobacteriota;c__Coriobacteriia;o__Coriobacteriales;f__Coriobacteriaceae;g__Collinsella 0.025594808940158614 0.024514264304447895
d__Bacteria;p__Firmicutes;c__Clostridia;o__Lachnospirales;f__Lachnospiraceae;g__Anaerostipes 0.01411061901328664 0.03832360969865653
d__Bacteria;p__Firmicutes;c__Bacilli;o__Erysipelotrichales;f__Erysipelatoclostridiaceae;g__Erysipelotrichaceae_UCG-003 0.017252034195076732 0.008456885939089012